U.S. Department of Transportation
Federal Highway Administration
1200 New Jersey Avenue, SE
Washington, DC 20590
202-366-4000
Federal Highway Administration Research and Technology
Coordinating, Developing, and Delivering Highway Transportation Innovations
 |
| This report is an archived publication and may contain dated technical, contact, and link information |
|
Publication Number: FHWA-RD-01-163 Date: March 2002 |
Previous | Table of Contents | Next
Physical attack mechanisms are related either to cyclic freezing and thawing of the concrete and/or the application of chemical deicers to keep the pavement surface free of ice. The three types of physical attack mechanisms considered here are freeze-thaw deterioration of the paste, deicer scaling/deterioration, and freeze-thaw deterioration of aggregate.
Freeze-Thaw Deterioration of Paste
When moist concrete is exposed to alternating cycles of freezing and thawing, internal deterioration can result. As this deterioration accumulates, it is referred to as freeze-thaw damage. Damage related to freezing and thawing can occur in both the cement paste and aggregate phases of concrete. It has long been recognized (Powers 1945) that damage to the cement paste phase can occur internally or at the surface. The issue of surface scaling is addressed later in the discussion of deicer scaling/deterioration, whereas susceptibility of aggregates to freeze-thaw damage is described in the section on aggregate freeze-thaw deterioration. This section focus exclusively on the damage that can occur to the cement paste phase internally due to repeated cycles of freezing and thawing.
Paste freeze-thaw damage in concrete is considered to be a physical phenomenon arising from excess internal pressures resulting from the freezing action of water. Currently, a general consensus on the exact mechanism(s) responsible for the generation of these excessive internal pressures does not exist. The most widely accepted theories consider either hydraulic or osmotic pressures (or a combination of the two) to be the primary causes. The role of entrained air voids is a central element in protecting the paste from damage in both of the theories. It is generally agreed that the magnitude of these internal pressures is dependent on the concrete pore structure, moisture content, pore water chemistry, rate of freezing, and/or length of the freezing cycle.
The temperature at which water will freeze is a function of the size of the pore in which it is contained and the concentration of dissolved species in the water. An excellent review of the literature related to these phenomena is provided by Marchand et al. (1994). Because of physical and chemical surface interactions between water and the surfaces of hydrated cement, the smaller the pore size, the lower the temperature required to cause freezing. Because of their small size, the water held in the gel pores will not freeze at the temperatures to which concrete is exposed in a natural environment. Because the pore water solution in a hydrated cement paste contains varying levels of sodium and potassium alkalis, the water in the capillary pores tends to become supercooled (in which it remains in a liquid state at temperatures well below 0°C) before freezing. This effect is magnified if deicing salts are also present.
Powers (1945) first attributed freeze-thaw damage to excessive hydraulic pressures resulting from the expansion of ice. It was proposed that as ice gradually forms at discrete sites in a saturated capillary pore system, the resulting 9 percent volume expansion causes the surrounding unfrozen water to be expelled away from the freezing sites. Depending on the nature of the pore system, excessive internal stresses can be generated by hydraulic pressures resulting from resistance to this flow. The pressurized water moving away from the freezing sites would find relief at the air voids, where it could then presumably freeze without causing damage.
Based on experimental results, Powers recognized that the spacing between voids, rather than the total volume of air, was the better measure of resistance to freeze-thaw damage. Building on this, he developed equations that provided an average measure of the distance that water must travel to reach the surface of an air void (Powers 1949). He proposed the adoption of a void-spacing factor, now known as the Powers spacing factor, as the basis of protecting the paste from freeze-thaw damage. It is interesting to note that this pioneering work based on the hydraulic pressure theory still forms the primary basis of specifying freeze-thaw resistant concrete (see ASTM C 457).
More recent theories (Powers 1975) consider osmotic potential to be the primary cause of excess internal stresses. As previously mentioned the temperature at which water will freeze in concrete is a function of the alkali concentration as well as the pore size in which it is contained. Freezing will only occur when the temperature becomes low enough to allow ice to form at the existing alkali concentration. Because of their relatively large size, air voids are likely initial freezing sites and as the pore water solution freezes, only pure water forms the ice. Thus, the remaining unfrozen liquid at the freezing sites becomes a more concentrated alkaline solution. The less concentrated alkaline solution in the surrounding paste is then drawn to the freezing sites to maintain thermodynamic equilibrium. The driving force for the movement of this solution is a function of the alkali concentration gradient. As the unfrozen solution at the freezing sites is diluted by the infusion of surrounding water, additional ice growth occurs. This progressive ice formation can occur at any solute concentration, including zero and is referred to as ice-accretion (Powers 1975). Recent experimental studies have provided some support for this mechanism (Wang et al. 1996).
This process of pore water moving from the capillary system to contiguous air voids will continue until one of two possible conditions prevails. If adequate air-void space exists, sufficiently distributed throughout the paste, all of the freezable water will eventually diffuse to the freezing sites inside the air voids, eliminating any further fluid flow. This is desirable, since the resulting total absence of freezable water in the surrounding capillary pore system eliminates the possibility of paste frost damage. The other possible outcome is that the air void space is inadequate to accommodate all of the surrounding unfrozen water. If this occurs, osmotic pressures will increase due to the remaining differences in alkali concentrations between the solution surrounding the ice-filled voids and the bulk solution within the capillary pores. Pressures of any kind, whether caused by loads, hydraulic forces, or osmotic forces, that approach or exceed the tensile strength of the hardened cement paste will naturally cause damage. Also, if the air-void volume is inadequate, freezable water will remain in the capillary pores. This freezable water in the capillary pore system is susceptible to ice crystal growth at sufficiently low temperatures. Although extreme temperatures are required to freeze water entrapped in capillary porosity, excessive hydraulic pressures may then result as unfrozen water is forced through the pore structure due to ice expansion. Also, if the rate of temperature drop is too fast to allow all of the water to diffuse to the air voids, damage may occur.
Deicer Scaling/Deterioration
Deicer Scaling/deterioration is typically characterized by scaling or crazing of the slab surface due to the repeated application of deicing chemicals. Although the exact causes of deicer scaling are not known, it is commonly believed to be primarily a physical attack. But some recent studies suggest that chemical degradation of the cement paste may also be occurring, resulting in dissolution of calcium hydroxide, coarsening of the concrete pore system, and potentially the formation of deleterious compounds. Both the physical and chemical deterioration mechanisms are discussed below.
As mentioned, while the exact causes of deicer scaling are not known, it is commonly thought to be primarily a form of physical attack, possibly resulting from a combination of factors (Mindess and Young 1981; ACI 1992a; Marchand et al. 1994; Pigeon 1994; Pigeon and Plateau 1995). Some researchers have stated that the presence of deicing chemicals, particularly salts, can magnify or amplify the same mechanisms that lead to freeze-thaw deterioration of the paste as discussed previously (e.g., hydraulic and osmotic pressures) (Ghafoori and Mathis 1997). Contributing to this effect is the fact that pore water containing a relatively small amount of dissolved salts is more easily absorbed into capillary pores, resulting in increased saturation of the concrete (Pigeon and Plateau 1995, Ghafoori and Mathis 1997).
In addition to the hydraulic and osmotic pressure theories discussed in the paste freeze-thaw section, another physical theory of why the application of deicing salts can produce concrete scaling is that of salt crystallization within the pores (Hansen 1963). It is hypothesized that the pore solution can become supersaturated as wetting and drying cycles concentrate salts in the pores (Harnik et al. 1980; Mindess and Young 1981). Once crystallization begins, salt molecules are drawn out of smaller pores into the larger pores, inducing potentially harmful crystallization pressures (Ghafoori and Mathis 1997).
It is apparent from the literature review that the physical mechanisms of deicer scaling are not completely understood. As stated by Pigeon (1994), deicer “scaling is a much more complex problem than frost induced internal cracking for many reasons, but probably in good part because it is related to the microstructure of the very surface layer or ‘skin’ of concrete.” Little is really known about the microstructure of this “skin,” except that it is composed predominantly of paste and that it is easily affected by poor finishing or curing practices. Because of this, it is subjected to high humidity gradients, drying, and microcracking and possibly an altered pore structure that is more susceptible to freeze-thaw damage (Pigeon 1994).
Although most research suggests that salt scaling is primarily a physical deterioration mechanism, some researchers have suggested that more attention should be paid to the chemical interaction between the deicing salts and cement paste hydration products (Marchand et al. 1994). Recent research indicates that the chemical interaction between deicers and concrete may not be as wholly benign as much past research suggests.
One major chemical degradation mechanism resulting from the long-term application of the popular chemical deicer sodium chloride (NaCl) is the dissolution of calcium hydroxide (Ca(OH)2). The dissolution equation is stated as follows (Marchand et al. 1994):
|
2NaCl + Ca(OH)2 à CaCl2 + 2NaOH |
(A-1) |
It is suggested that the dissolution process results in increased porosity at exposed surfaces, increasing the permeability of the concrete. This in turn increases the amount of water available and that which will freezes at 0°C due to increased pore size (Marchand et al 1994). One recent study conducted by the Michigan Department of Transportation (MDOT) adds credence to this theory. Muethel (1997) confirmed through laboratory analysis that depletion of calcium hydroxide led to an increase in permeability and reduced alkalinity of the concrete in the vicinity of cracks and joints. The reduction in the concrete pH contributed to corrosion of reinforcing mesh in the jointed reinforced pavements.
A subsequent reaction may occur in which the soluble calcium chloride reacts with aluminate phases in the paste to create chloroaluminate crystals according to the following equation (Marchand et al 1994):
|
CaCl2 + C3A à C3A· CaCl2·10H2O |
(A-2) |
It appears that this reaction would be expansive, although no citation was found in the literature regarding the expansive pressures that would be exerted, or whether it would be damaging in and of itself. Buck (1985) mentions that chloroaluminate preferentially replaces ettringite when salt is present. Buck also discusses that chloroaluminate formed during initial hydration in the presence of calcium chloride could later convert to ettringite if the solution becomes rich in sulfate ions.
Collepardi et al. (1994) cited a number of studies that have concluded that CaCl2, another common deicer, is associated with a deleterious chemical reaction with concrete. The chemical attack is accompanied by the formation of a hydrated calcium oxychloride according to the following reaction:
| 3Ca(OH)2 + CaCl2+ 12H2O à 3CaO · CaCl2 · 15H2O | (A-3) |
The reaction proceeds most efficiently at temperatures just above freezing (5°C to 10°C), with rapid formation of hydrated calcium oxychloride. This reaction is considered to be disruptive to the concrete matrix because of the hydraulic pressures generated. Collepardi et al. (1994) speculate that the damaging nature of this reaction has been masked by corrosion of reinforcing steel and freeze-thaw deterioration to the paste, but state that the chemical degradation that occurs is very detrimental. Collepardi et al. (1994) cite experimental evidence, based on decreasing compressive strength, suggesting that severe deterioration occurred in non-air entrained concrete exposed to CaCl2 deicers even though there was no steel to corrode nor was the concrete subjected to temperatures below freezing.
It would be expected that factors leading to an increased permeability would be more prevalent at exposed concrete surfaces. In addition to contributing to surface scaling, concrete faces along joint or crack walls would also be adversely affected. A number of studies appear to support this hypothesis. The previously cited study conducted by MDOT observed that CH leaching was pronounced in concrete cores obtained at transverse crack locations in jointed reinforced concrete pavements from three different State routes (Muethel 1997). Visual surveys of the concrete pavement surface revealed localized deterioration and staining, while laboratory analysis confirmed depletion of CH, increased permeability, and reduced alkalinity of the concrete.
A study conducted for the Ohio Department of Transportation (ODOT) investigated the joint deterioration observed in many northern tier States (Munoz and Chou 1994; 1996). Termed "coning," this deterioration is characterized by concrete degradation at joints that progresses from the bottom of the slab upward. The exact cause of this deterioration is unknown, but it is speculated that more than one distress mechanism might be at work. Munoz and Chou state that dowel baskets may interfere with proper concrete consolidation, resulting in weaker concrete and increased porosity and permeability. This contributes to a chemical attack mechanism, with dissolution of the concrete considered the primary culprit. Munoz and Chou (1994; 1996) explored the dissolution mechanism, stating that both the presence of dissolved salts and the velocity of solvent flow through the concrete are important factors. It is speculated that concrete at the joints has increased permeability, with accompanying relatively high solvent velocity. Additionally, potentially high CO2 content in the solution from melted snow and other factors may lead to an increased solubility of the concrete.
Another potential detrimental effect of the application of chemical deicing salts is increased alkali–silica reactivity. Nixon and Page (1987) report that salt contamination of concrete, whether through the incorporated aggregate or application of deicing salts, " could be expected to increase the alkalinity of the pore solution and hence the likelihood of damaging alkali silica reaction." The mechanism cited is the combination of sodium chloride with calcium hydroxide and tricalcium aluminate, precipitating calcium chloroaluminate or chloro-sulphoaluminate gel as previously described. They state that in cases where sodium chloride is made available through deicer applications, the effect is to produce a zone of reduced alkalinity near the exposed concrete surface, an occurrence consistent with the observations of Muethel (1997).
Aggregate Freeze-Thaw Deterioration
Freeze-thaw deterioration of aggregate is associated with the freezing and thawing of susceptible, coarse aggregate particles in the concrete. The resulting pavement distress is commonly referred to as D-cracking. Aggregates identified as being freeze-thaw susceptible either fracture as they freeze and then dilate resulting in cracking of the surrounding mortar, or they allow for rapid expulsion of water during freezing contributing to dissolution of soluble paste components at the aggregate-paste interface. Key aggregate properties related to freeze-thaw susceptibility are aggregate size, pore size distribution, and strength (Mindess and Young 1981). Most susceptible aggregates are of sedimentary origin (e.g., cherts, sandstones, shales, limestones), can be calcareous or siliceous, and can be gravel or crushed rock (Neville 1996).
Two different mechanisms are associated with aggregate related freeze-thaw damage, including damage to the aggregates themselves and/or damage to the adjacent paste system. Powers’ hydraulic pressure theory is generally considered to provide a reasonable description of the actions taking place inside aggregate particles during freezing (Pigeon and Plateau 1995). In a critically saturated aggregate, excessive pressures can develop due to the volume increase associated with ice formation. The expansion associated with ice formation may be accommodated by either elastic deformation and/or the expulsion of unfrozen water. If an aggregate is less than critically saturated, or can expel water to its surroundings, internal hydraulic pressures result as unfrozen water is forcibly displaced by ice formation. The hydraulic pressures are greater when this flow is through smaller sized pores and along longer flow paths (Winslow 1994). The tensile capacity of an aggregate can be exceeded when either the expansion due to ice formation cannot be accommodated by elastic deformation or the hydraulic pressures become excessive. In other cases, the expulsion of water from sound aggregate particles during a freezing event can result in damage to the surrounding paste phase.
It is fairly well established that the aggregate pore system is the most important factor contributing to its freeze-thaw durability (Schwartz 1987; Winslow 1994; Pigeon and Plateau 1995). Two related elements of the pore system that are of particular interest are the porosity and pore size distribution. Aggregate porosity is the ratio of the surface accessible pore volume to the total aggregate volume. Porosity can be easily determined, providing a measure of the total volume of water that can be contained within a fully saturated aggregate. Pore size is a measure of the physical dimensions of the various elements of the accessible pore volume. It is a much more difficult characteristic to determine, but provides an important indication of an aggregate’s potential to become critically saturated. It also provides a measure of its permeability or resistance to fluid flow during a freezing event.
Perhaps the single most significant pore characteristic is the volume of the total porosity contained within a range of pore sizes. This parameter has been empirically related to aggregate freeze-thaw durability (Kaneuji 1980). The exact limit of detrimental pore sizes is not definitively established, but has been reported to be in the range of under 0.10 mm up to 5 mm (Kaneuji et al. 1980; Marks and Dubberke 1982; Shakoor and Scholer 1982). Elements of the pore system smaller than the lower limit are not considered detrimental for one of two reasons. Either their size is such that the water in them will not freeze at normal winter temperatures or the contained volume of water is so small that it does not contribute significantly to stresses in either the aggregate itself or the surrounding paste. Pore systems with sizes greater than the upper limit are not considered detrimental to the aggregate because either they do not become critically saturated or their permeability is so high that they do not generate excessive hydraulic pressures. Aggregates with a preponderance of pore sizes within the detrimental size range are believed to have a higher potential to become critically saturated and/or develop excessive hydraulic pressures due to low permeability.
Because internal hydraulic pressures are directly proportional to the length of the flow path, reducing the size of the aggregate can reduce the magnitude of these internal pressures. Reducing the aggregate size can also result in a smaller volume of expelled water per unit surface area. Experimental studies have confirmed an increased freeze-thaw durability of susceptible aggregates with a reduction in maximum particle size (Stark and Klieger 1974). The maximum particle size required to ensure a freeze-thaw durable aggregate varies greatly because it depends on the characteristics of the aggregate pore system, including the porosity and the pore size distribution (which will affect the degree of saturation and permeability), along with the mechanical properties of the aggregate. Additionally, the properties of the surrounding paste system influences the value of this maximum size. While reducing the maximum particle size may improve the freeze-thaw performance of some aggregates, it also typically results in an increased cement paste/mortar volume. In addition to being more expensive, a higher paste volume can increase drying shrinkage and result in reduced aggregate interlock at joints and across structural cracks. It also will increase the total alkalinity per unit volume of concrete and thus may contribute to alkali-aggregate reactivity (Leming 1996). The fine aggregate fraction is likely unassociated with aggregate-related freeze-thaw deterioration due to their size.
Alkali–Silica Reactivity
Alkali–silica reactivity (ASR) is a distress caused by undesirable chemical reactions between alkalis in the cement paste (Na2O and K2O) and the reactive siliceous components of susceptible aggregates. The product of the reaction is expansive in the presence of moisture, destroying the integrity of the weakened aggregate particle and the surrounding cement paste (Mindess and Young 1981). The reaction product is often referred to as a gel, which is a misidentification in some instances.The term gel implies an amorphous structure when in fact, often, the product is crystalline. ACI (1998) provides an in-depth description of this chemical attack mechanism.
With time, the alkalis in the paste pore solution may react with certain forms of reactive silica contained in the aggregate forming the alkali-silica reaction product. The most reactive forms of aggregate are strained quartz, amorphous silica, cryptocrystalline quartz, chalcedony, and chert. If water is available, the reaction product will draw it out of the surrounding cement paste, causing swelling, exerting potentially damaging tensile stresses from 4,100 to 11,000 kPa within the cement matrix (Farny and Kosmatka 1997). These stresses may also be large enough to cause bond and shear failures between concrete and reinforcement (CCAA 1996).
The penetration of hydroxyl ions into the siliceous aggregates breaks the silicon-oxygen linkages within the silica tetrahedra. The resultant negatively charged silicon hydroxide ions are electrically balanced through interaction with the sodium and potassium ions in the pore solution, forming the new reaction product (Harrison et al. 1987). Swelling occurs as the forces of attraction between the polar water molecules and the alkali–silicate ions results in chemically bound water in the solid reaction product.
Factors that affect the rate at which ASR develops include the amount and type of siliceous materials present, the concentration of alkalis in the pore water solution, and the amount of moisture accessible to the gel product. At higher temperatures, gel expansion is greater but stabilizes earlier (CCAA 1996). Certain rock types containing silica are more reactive than others. In addition to the minerals named previously, the following rock types are known to be potentially reactive: rhyolite, dacite, latite, andesite, tuffs, shale, slate, sandstone, siltstone, quartzite, granites, grano-diorites, and granite gneisses. This list is by no means complete, but it shows that a large number of rocks may be reactive and deleterious to concrete (Farny and Kosmatka 1997). In general, aggregates containing crystalline silica are stable and those with amorphous or very fine grained silica are reactive. Also, aggregate with large surface areas or many lattice defects are more susceptible to ASR.Thus, the potential reactivity of an aggregate is a function of both its degree of crystallization and the amount of energy stored in the crystal structure (Leming 1996).
The concentration of alkali in the pore solution is the second factor affecting the rate of ASR development. This factor is affected by two main sources: the alkali content of the cement and alkali addition from other sources, both internal and external. The alkali content of cement is normally expressed as Na2O equivalent alkalis (Na2O percent + 0.658 percent K2O). Low alkali cement is typically considered to have an Na2O equivalent of 0.6 percent or less. But it is noted that the total alkalis from the cement in a given concrete is the product of the Na2O equivalent and the cement content. Thus, a mixture made with low alkali cement may in fact have high total alkali content if a large amount of cement is used. For this reason, European standards specify a limit of 3 kg/m3 on the total alkali content per unit volume of concrete (Leming 1996). Internal sources of alkalis other than the cement include mineral or chemical admixtures, aggregates, and even the mixing water. External sources of alkalis include deicing salts, seawater, groundwater, and water from industrial processes that permeate the concrete (Farny and Kosmatka 1997).
It is noted that a highly alkaline pore solution is not necessary for ASR to develop. Aggregate composed of highly reactive silica may suffer ASR even if the pore solution has a relatively low concentration of alkalis. Alternately, if the alkali concentration of the pore solution is high enough, it can break the strong silica bonds present in less reactive aggregates resulting in deleterious ASR (Farny and Kosmatka 1997). ASR has also been known to develop in areas of localized high alkalinity even if the alkalinity of the rest of the pore solution is low.
The third factor affecting the expansion of the ASR reaction product is the amount of accessible moisture. It has been found that an internal relative humidity of at least 75 to 80 percent is needed for deleterious ASR to occur (Farny and Kosmatka 1997; CCAA 1996). This internal relative humidity is common in pavement concrete. It is still unclear whether continuous saturation or cycles or wetting and drying cause more ASR-associated damage (Farny and Kosmatka 1997; CCAA 1996).
Alkali–Carbonate Reactivity
As discussed in the previous section on ASR, the pore solution of concrete is highly alkaline, primarily a result of the sodium and potassium ions released during cement hydration. Excess alkalis are also derived from admixtures, mixing water, fly ash, GGBFS, silica fume, and external sources (CCAA 1996). The exact mechanism of ACR is still debated, but it is believed that these alkalis react with the carbonate aggregates in a process called dedolomitization, a breaking down of the dolomite to form brucite, calcium carbonate, alkali carbonate, and alkali hydroxide (Farny and Kosmatka 1997; ACI 1998). The type of aggregates susceptible to this type of reaction are typically dolomitic limestone that consist of a fine-grained matrix of calcite and clay in which larger crystals (20-80 um) of euhedral dolomite rhombohedra are suspended (Ozol 1994). Dedolomization can be represented by the following equation in which M represents potassium, sodium, lithium or other alkali element (Ozol 1994):
| CaMg(CO3)2+ 2MOH à Mg(OH)2 + CaCO3 + M2CO3 | (A-4) |
The alkali carbonate produced in this reaction may then react with Ca(OH)2 produced through normal cement hydration to regenerate the alkali hydroxide, an example of which is illustrated in the following equation (Ozol 1994):
| Na2CO3+ Ca(OH)2 à 2NaOH + CaCO3 | (A-5) |
Although the starting and end products of this reaction are well documented, the expansion portion of the reaction is still being debated. There are two main theories on how the expansion occurs in concrete. The first theory suggests that the expansion is caused by migration of alkali ions and water molecules into the restricted space of the fine-grained matrix surrounding the dolomite rhomb. Typically, the alkali reactivity of carbonate rocks is not dependent on the clay mineral composition (Farny and Kosmatka 1997). This theory argues that expansion is caused when clay, exposed by dedolomitization, attracts and absorbs water.
The second theory is that the growth and rearrangement of the de-dolomitization products, especially brucite, exerts pressure as it crystallizes. This pressure from the crystallizing products causes expansion (Farny and Kosmatka 1997; Gillott 1995). Leger et al. (1995) and Nixon and Page (1987) suggest that the first theory is correct and that an expansive reaction occurs with clay minerals. On the other hand, Mingshu et al. (1994) added credence to the second theory by showing that de-dolomitization itself causes expansion. Ozol (1994) suggests that both theories might be correct, stating that ACR expansion is a result of a combination of both mechanisms. The literature suggests that more research is required before the absolute mechanism of deterioration is fully understood.
Regardless of the mechanism, it is known that for the ACR reaction to occur, three criteria must be met. First, the content of reactive aggregate must be in excess of a critical value. Second, there must be sufficient alkalis present in the pore solution of the concrete to initiate a reaction. Lastly, sufficient moisture must be available so that the reaction may proceed (Leger et al. 1995). As with ASR, ACR requires a source of moisture in order to have deleterious consequences. Thus ACR is not common if the concrete has an internal relative humidity less than 75 percent (CCAA 1996).
Swamy (1994) identified three aggregates with which carbonate reactions commonly occur. These aggregates are calcitic limestones, dolomitic limestones, and fine-grained dolomitic limestone aggregates containing interstitial calcite and clay. Farny and Kosmatka (1997) outline aggregates that have potential for ACR in the following manner:
In ACI (1998), the concept of early expanders and late expanders is discussed. The difference between the late and early expanders is reflected in the bulk composition and internal textural restraint. Late expanders typically have a higher acid insoluble residue (21 to 49 percent) and a higher percent of dolomite present (75 to > 90 percent) of total carbonate (ACI 1999). The degree to which reactive carbonate rocks will cause expansion in concrete is related to the restraint imposed by the concrete, the volume of dolomite in the rock, and the internal textural restraint of the carbonate rock (Ozol 1994). Ozol (1994) also reports that the expansion of concrete containing alkali reactive carbonate rocks is promoted by increasing coarse aggregate size, moisture availability, higher temperature, high alkali content of concrete, high pH of the liquid phase in cement pores, high proportion of reactive stone in coarse aggregate, and lower concrete strength.
Sulfate Attack
As with most other MRD mechanisms, the basic mechanism of sulfate attack has been thoroughly investigated but not fully understood (DePuy 1994). The first step in the process is that sulfate ion become available within the concrete pore solution, either through diffusion of sulfate ions into the concrete from the outside or through internal sources. The permeability of the concrete and the diffusion coefficient of the sulfate ions control this process. Microcracking, which may result from the sulfate attack or other causes, increases the permeability of the concrete, leading to acceleration of the damage (Mindess and Young 1981). However, Hughes (1985) found that susceptibility to sulfate attack was not simply a function of water permeability. He proposed that resistance to sulfate attack is related specifically to the ability of the sulfate ion to diffuse through the pore structure. This is not only related to permeability, but also to the ability of the sulfate ions to then gain access to the susceptible minerals. This in turn is related to the entry size of the pores associated with those minerals. Gowripalan et al. (1993) cite several studies that conclude that the flow of a fluid through concrete is related to the larger capillary pores and not the total porosity; Scrivener (1996) found that the interfacial transition zones around aggregate particles are an important factor in the permeability of the concrete. Thus, the volume of larger pores and the characteristics of the interfacial zone are correlated with the concrete’s resistance to sulfate attack.
Once sulfate ions have diffused into the pore system, the conversion of sulfoaluminates to expansive ettringite, also known as calcium trisulfoaluminate (3CaO·Al2O3·3CaSO4·32H2O) can occur. This is the cause of most of the disruption of the concrete matrix due to sulfate attack (ACI 1992a). The exact mechanism of this disruption is still controversial. Mehta and Monteiro (1993) state that there is general agreement that expansion resulting from sulfate attack is related to ettringite formation that occurs when sulfate ions attack the calcium hydroxide and alumina-bearing phases of a hydrated portland cement paste. Therefore, many researchers have correlated the sulfate resistance of cement to its C3A content because it controls the amount of sulfoaluminate hydration products that will form in the paste. Mindess and Young (1981) agree with this, stating that the susceptibility of concrete to sulfate attack is related to how much ettringite can form, which relates back to the amount of C3A in the cement.
Mehta and Monteiro (1993) state that portland cements with more than 5 percent potential C3A will contain most of the alumina as monosulfate hydrate (C3A·CaSO4·H18). If the C3A content is more than 8 percent, the hydration products will also contain C3A·CH·H18. In the presence of calcium hydroxide and sulfate, both of the alumina-containing hydrates are converted to the high sulfate form of ettringite, according to the following reactions:
| C3A· C |
(A-6) |
|
C3A· CH ·H18 + 2CH + 3 |
(A-7) |
Bickley et al. (1994) present the ettringite formation reaction in the following manner:
| 6Ca2++ 2Al(OH)4- + 4OH- +
3 |
(A-8) |
They state that the required Al(OH)4- can be supplied by C3A or C4AF in the un-reacted clinker, but is more likely from the monosulfoaluminate (3CaO·Al2O3·CaSO4·18H2O). Wolter (1997) states that ettringite can also form from gypsum reacting with the C3A. Calcium monosulfoaluminate (3CaO·Al2O3·CaSO4·12H2O) forms if the concentration of sulfate is below the point at which ettringite is stable (DePuy 1994; Chatterji 1969). In a higher sulfate solution, the calcium monosulfoaluminate is converted to ettringite (DePuy 1994). In both of these forms, part or all of the Al3+ ions can be replaced by Fe3+ ions (Chatterji 1969).
Although there is general agreement that sulfate-related expansions in concrete are associated with ettringite, how the ettringite causes expansion is still a subject of controversy. Exertion of pressure by growing ettringite is one theory, supported by Wolter (1997) and Lafuma (in Chatterji 1969). Lafuma’s theory is that ettringite formation can take place through two different mechanisms. If the liquid phase containing sulfate ions is saturated with CH, then ettringite is formed in a solid-state reaction between C3A and the SO42- and Ca2+ ions in solution. This reaction causes a volume increase of about 815 percent on the basis of C3A volume. If the liquid phase is unsaturated with respect to CH, the reaction occurs in the liquid phase and ettringite formation occurs only in the existing pores in the structure without causing expansion (Chatterji 1969).
Swelling due to adsorption of water in an alkaline environment by poorly crystalline ettringite is another theory supported by Mehta (1991) and Wolter (1997). Mehta (1991) cites a study that provides evidence of swelling of microcrystalline ettringite. Additionally, Wolter (1997) speculates that ettringite needs a high pH environment in order to crystallize. This is based on observations that in concrete with extensive ettringite, there is no ettringite where carbonation has occurred. Wolter (1997) speculates that the ettringite may dissolve and move to an area of higher pH and recrystallize as secondary ettringite in available void space.
The formation of thaumasite (CaCO3·CaSO4·CaSiO3·15H2O), a mineral that is structurally similar to ettringite, may also contribute to sulfate attack. It has been speculated that thaumasite is not often identified in concrete because it is often misidentified as ettringite due to their similarity in structure and appearance (Rodgers et al. 1997). Berra and Baronio (1987) describe it as a complex sulfate salt, a heavily hydrated triple compound of calcium metasilicate, calcium sulfate, and calcium carbonate. It is similar to ettringite, except that silica is substituted for alumina (Crammond 1985; Berra and Baronio 1987). DePuy (1994) states that it may form as a conversion product of ettringite with carbonation and silicon substitution, or directly when there is a supply of alumina, calcium silicates or free silica gel, sulfate, and carbonate.
Thaumasite forms preferentially at low temperatures because of the high solubility of calcium salts at low temperatures (DePuy 1994). It may also form due to the higher solubility of CO2 at cold temperatures, which favors a carbonation reaction necessary for thaumasite formation (Bickley et al. 1994). At 5°C, thaumasite is a hundred times less soluble than it is at 20°C, which may partially explain some cold temperature deterioration (Crammond and Halliwell 1995). For this reason, it is thought to be more prevalent in colder climates such as the northern U.S. and Canada (Rodgers et al. 1997).
Unlike ettringite formation, the formation of thaumasite is dependent on the more abundant supply of CSH and not on the C3A content. This means that the deterioration is not affected nor limited by C3A content, and can proceed until there is no binding agent left, completely destroying the concrete. Thaumasite formation has been found to occur more readily with dolomite aggregates, attributed to the dedolomitization of the aggregate that provides additional CaCO3 (Crammond and Halliwell 1995). Crammond and Halliwell (1995) suggest that all types of PCCs containing finely divided carbonate rock dust as a source of carbonate ions may be potentially vulnerable to thaumasite attack, based on very limited evidence.
In addition to formation of ettringite or thaumasite, sulfate attack also occurs due to the formation of gypsum. This mechanism is commonly referred to as gypsum corrosion. Gypsum corrosion only contributes directly to the deterioration of the concrete at sulfate concentrations above 1,000 ppm and is only of secondary importance until sulfate levels exceed 4,000 ppm (Mindess and Young 1981). Gypsum (CaSO4·2H2O) formation takes several forms, depending on the form of the attacking sulfate. With relatively small concentrations of sulfate ions, the nature of the sulfate ion does not matter. But as ion concentration increases, the nature of the ion has an influence. At lower concentrations (SO4 < 1000 mg/L), sodium sulfate reacts to form ettringite, and at higher concentrations, it produces gypsum (DePuy 1994). For sodium sulfate gypsum corrosion, Mehta and Monteiro (1993) gives the following reaction:
| Na2SO4 + Ca(OH)2 + 2H2O àCaSO4 ·2H2O + 2NaOH | (A-9) |
In this case, the formation of sodium hydroxide as a byproduct of the reaction maintains the high alkalinity of the system, protecting the stability of the CSH phase.
External Sulfate Attack
The most common source of external sulfates is from soils or groundwater. Impurities in deicing chemicals can also provide a ready source of external sulfates. This has been labeled deicer distress and has been gaining attention in some northern States. Sulfates are a common impurity in many naturally occurring deicing salts. Standard NaCl deicing salts are required to be as little as 92 percent pure, allowing for up to 8 percent impurities. These impurities are often gypsum (CaSO4·H2O) and anhydrite (CaSO4) (Wolter 1997). One research project found that natural rock salt may contain as much as 4.0 percent calcium sulfate, which increases in solubility in a chloride solution (Pitt et al. 1987). The amount and increased solubility of the calcium sulfate was found to be sufficient to cause deterioration to concrete mortar.
According to Wolter (1997), exterior concrete exposed to moisture and a natural deicing agent with gypsum or anhydrite impurities under freeze-thaw conditions can be potentially affected by deicer distress as the gypsum impurity in the deicer reacts with the C3A to form ettringite. As Munoz and Chou (1996) state, the gypsum reacts with the hydrated calcium sulfoaluminates and calcium aluminates as described previously. Deicer distress is different from a standard sulfate attack in that the gypsum is introduced directly, without the reaction between the sulfate and calcium hydroxide (Wolter 1997). Under these conditions, it is speculated that the presence of gypsum leads directly to ettringite formation as described above.
In addition to deleterious expansion, the ettringite may also go into solution, recrystallizing within the pore space and air void system of the concrete. The process of void infilling is incremental, as air voids would only become partially filled with water as it moves from the paste to relieve hydraulic/osmotic pressures developed during freezing. As the water enters the air void, it forms ice crystals on the pore wall. When the crystals melt, secondary products are deposited on the pore walls (Niemann and Lehtonen 1997). Over time, incremental deposition of ettringite and other secondary deposits on the walls of air voids may completely fill the interstitial pore space and air-void system, leaving the concrete vulnerable to freeze-thaw deterioration.
The incremental filling of the voids and resulting vulnerability of air-entrained concrete to freeze-thaw damage has been verified through an extensive study conducted in Sweden (Niemann and Lehtonen 1997). Over 1,500 thin sections were petrographically analyzed. Scanning electron microscopy and energy dispersive x-ray microanalyses were used to study the deposits as well as judge the extent of void infilling. This study found that void infilling was progressive, with increasing void volume being filled over time and smaller voids were filled first. This study also demonstrated a decrease in freeze-thaw durability with an increase in volume of voids filled. The density of the material filling the voids approached that of the aggregate, and the study concludes that these voids were no longer able to protect the concrete against freeze-thaw damage. Similar results were found in a German study (Bollmann and Stark 1996).
Dubberke (1993) gives a similar, but slightly different, explanation for the deterioration of Highway 520 in Iowa. He theorizes that the pores became filled with "amorphous ", non-crystalline ettringite. He suggests that expansion could occur from the generation of the amorphous ettringite paste in small pores (<50 microns), first in high moisture areas such as the bottom of the slab and near joints. This causes differential expansive forces between the top and bottom of the slab, leading to cracking. The cracking occurs in the weakest areas, such as the vibrator trails that lack coarse aggregates and have a relatively high w/c ratio. However, it has been noted by some that the expansive pressures generated by secondary deposition of ettringite in available void space is insufficient to cause damage to hardened concrete (Scrivener 1996; Johansen et al. 1993).
It must be noted that finding ettringite in microcracks in the paste is in itself not sufficient evidence to link the distress mechanism to external sulfate attack. Ettringite is a common component of hydrated cement paste and the question arises as to whether the observed cracking was caused by the ettringite formation or if secondary ettringite merely formed in the available space (DePuy 1994). This makes diagnosis of external sulfate attack difficult in some cases.
Internal Sulfate Attack
The term internal sulfate attack (ISA) is used to describe the deterioration of concrete resulting from sulfate attack where the source of the sulfate ions is from within the concrete itself. DePuy (1994) describes sulfates that may attack concrete. Potassium sulfate and sodium sulfate strongly react with PCC. Sulfates having very low solubility in water, such as barium and lead sulfate, which are common in some aggregates, are relatively harmless. Calcium sulfate is less aggressive than sodium sulfate, and magnesium sulfate is the most aggressive (Mielenz 1964). Although all of the reactions previously described in the discussion of external sulfate attack apply, and many sulfate phases can form within concrete, it is the formation of ettringite that is clearly the most important issue related to internal sulfate attack.
Ettringite is a normal meta-stable phase produced in the cement hydration process. It forms from the reaction of C3A or monosulfoaluminate with sulfate ions. The formation of ettringite by alteration of an existing solid phase is an expansive formation and, if the crystals are allowed to form while confined in the cement paste, a process of cracking will initiate within the paste.
To help establish a standard nomenclature on this issue, the American Concrete Institute's Committee 116 (Names and Definitions) is working on definitions for the various forms of ettringite. Preliminary definitions are presented in table A-1 (Erlin 1996a).
Delayed ettringite formation (DEF) and excess sulfate (EXS) are two types of internal sulfate mechanisims identified by Thaulow et al. (1996a). Secondary ettringite formation (SEF) is a third potential mechanism of internal sulfate attack. These have been increasingly identified as the cause of many cases of distress in PCC. DEF is most often associated with steam curing. At elevated temperatures (current research suggests a minimum temperature of 65 C to 80C [Scrivener 1996; Thaulow et al. 1996a; Klemm and Miller 1997]), primary ettringite will not properly form. After the concrete has cured and temperatures are reduced to ambient conditions, sulfates and aluminate phases in the paste may then react to form expansive ettringite, disrupting the concrete matrix. Because this phenomenon is most closely associated with steam curing, it is still speculative whether cast-in-place pavements can experience the temperatures necessary to produce DEF. EXS, on the other hand, appears to be a more likely mechanism for ISA. Possible sources of internal sulfate include either slowly soluble sulfates or sulfur compounds in the clinker or fly ash that only become available during continued long-term hydration. Another internal source of sulfates might be from aggregates (Johansen and Thaulow 1999). In these cases, the internal sulfate attack is not from DEF, but instead from excess sulfates in the concrete mixture, which result in paste expansion along similar lines as external sulfate attack.
Form |
Definition |
|
Ettringite |
A high sulfate calcium sulfoaluminate mineral (3CaO·Al2O3·3CaSO4·32H2O). |
|
Primary Ettringite |
Ettringite formed by reaction of sulfate and aluminate ions during early hydration of hydraulic cement either as a normal process for portland cement or as the expansive process for expansive cement. |
|
Secondary Ettringite (SEF) |
Ettringite formed by precipitation from solution of primary ettringite. |
|
Delayed Ettringite (DEF) |
Ettringite formed by reaction of sulfate and aluminate ions in concrete, mar, or grout that has hardened and developed its intended strength. If the sulfate is from an external source, the process is "sulfate attack;" if the sulfate is from within the concrete, the process is "delayed ettringite formation." |
Currently, there is no consensus among researchers about possible SEF distress mechanisms. Also, there is considerable debate about the internal sources of sulfate, with portland cement, aggregates, and fly ash all identified as possible sources. Based on the given definitions, it is evident that the composition of the cement may play a role in internal sulfate attack, but compositional variations are difficult to isolate due to the numerous producers, variations in production methods, and the range of raw materials and fuel sources used to produce portland cement. Other sources of sulfate, such as fly ash and aggregates, are being limited in many cases by strict specifications in an effort to control internal sulfate attack.
From the literature, it is almost impossible to separate DEF from EXS, particularly in nonsteam-cured structures such as pavements. As a result, these two will be treated together unless the mechanism at work is clearly identified. The basic mechanism of deterioration in DEF/EXS is the bulk expansion of the paste caused by reactions between solid phases within the cement paste and internally derived sulfate ions. In the DEF/EXS reaction, at least one of the reactants (i.e., usually aluminate or ferrite cement clinker phases or monosulfoaluminate) begins the reaction as a solid within the hardened cement paste and undergoes a solid-solid phase transformation with a significant increase in volume. This results in crack initiation. Mindess and Young (1981) give the following reaction for the conversion of tricalcium aluminate (C3A) and gypsum to ettringite:
| C3A + 3C |
Note that this reaction is a normal occurrence in the early hydration of portland cement. The crystallization pressure of ettringite forming from un-hydrated C3A is estimated as high as 240 mPa (35,000lb/in2) and is sufficiently large enough to cause cracking in concrete (Mindess and Young, 1981).
Portland cement clearly contains phases, such as tricalcium aluminate, that serve as the principal source of aluminate for ettringite formation. More importantly, although aggregates and external sources of sulfate can contribute significantly to the total sulfate in the concrete, portland cement is the primary source of the sulfate required to form ettringite (Erlin 1996b). For the most part, the constituents of portland cement have not changed over the years and cement producers argue that with modern quality control procedures, a more uniform product is now being produced than in the past. However, the sulfate content of portland cement has increased over the years. A study conducted by the PCA indicates that between the 1950s and 1994, the average sulfate contents in portland cement have increased almost 58% from 1.9% SO3 to 3.0 % SO3 for Type I cements (PCA 1996). Similar increases were noted for Type II cements.
At least part of this increase in sulfates is due to higher amounts of gypsum being added to address the more rapid reactivity of finely ground modern cements. But it has been hypothesized that some of this increase in sulfate content is a result of burning waste fuels. Such sulfate would be included in solution in the silicate phases of the cement clinker (Mielenz et al. 1995; Hime 1996, Wolter 1997). It is speculated that this sulfate is slowly soluble and would not contribute to the total sulfate available to form primary ettringite until coming out of solution at the latest stages of hydration. This hypothesis has gained acceptance due in part to a well-publicized case of distress in 400,000 steam cured cement railroad ties in the northeastern part of the United States (Mielenz et al. 1995). In this case, a team of consulting engineers determined that the majority of the ties failed as a result of DEF/EXS and that the sulfate source for ettringite formation was anhydrite phases and sulfate in solution in the cement clinker silicate phases. It is noted that this hypothesis is contested, and that the observed DEF had less to do with clinker sulfates than it did with curing practices, particularly high curing temperatures (Scrivener 1996).
Another point of contention is the classification method used to describe fly ash. Current methods (ASTM C 618) fail to adequately characterize fly ash that may be deleterious. Prescriptive tests that determine the soluble sulfate content and the soluble alumina content are needed (Dewey et al. 1996). The application of the former test is obvious, as knowing the total sulfate content in a mix is essential to predicting the formation of ettringite. In the latter case, the alumina component of the fly ash must be easily soluble to prevent alumina rich inclusions from forming in the CSH. If such inclusions form, they may be susceptible to sulfate attack and expansion (Gress 1997).
A key variable controlling DEF is the conditions under which the concrete is cured. In particular, the temperature of the concrete during curing appears to be an important factor influencing the development of delayed ettringite within that concrete. Concrete distress as a result of DEF is most common when the concrete is cured at elevated temperatures (i.e., steam curing) (Day 1992). As a matter of definition, Thaulow et al. (1996a) state that elevated temperatures are absolutely necessary for DEF to occur. The initial mechanism suggested for the occurrence of DEF at elevated temperatures is the thermal decomposition of primary ettringite formed during the early stages of hydration. After the concrete has cooled, delayed ettringite forms from the decomposition reaction products and causes the cement paste to expand, creating gaps around coarse aggregate particles. It is within these cracks that secondary ettringite may form, filling this newly created volume (Skalny 1996).
Although the high temperatures needed for DEF are normally associated with steam curing, some researchers have reported DEF at concrete curing temperatures obtainable during cast-in-place construction. The internal temperatures in pavements cured under high ambient temperatures may approach the critical temperature for DEF, which current research suggests is a minimum temperature of 65°C to 80°C, with many citing 70°C (Scrivener 1996; Thaulow et al. 1996a; Klemm and Miller 1997). It has been proposed that excess heat from the exothermic hydration process can increase the chances of DEF occurring (Hime 1996). Scrivener (1996) indicates that in the presence of excess sulfate, elevated temperatures (60 to 100°C) will increase the chance of DEF and that the ettringite that forms is microstructurally different than ettringite formed as a result of high sulfate content only. This research is consistent with field evidence that DEF is most often occurs in steam-cured concrete.
However, another researcher has shown that DEF is seen in nonsteam-cured concrete and that its microstructure is similar to ettringite formed in concrete cured at lower temperatures (Diamond 1996). Gress (1997) indicates that some cements are known to form delayed ettringite at temperatures as low as 45°C which is clearly attainable in slab-on-grade construction under slightly adverse weather conditions, especially if a high cement content mix is being used. These instances of internal sulfate attack may be more closely related to EXS and further research is required.
Internal sulfate attack by SEF is distinguished from that previously described under external sulfate attack through consideration of the source of the sulfate ions. If the source is from a concrete constituent or dissolved primary or delayed ettringite, it is considered to be from an internal source. If the sulfate source is from an external source, such as from groundwater or deicer impurities, the SEF would be considered as an external sulfate attack.
In SEF, the ions reacting are in the pore water and precipitate out of solution as ettringite into available spaces such as voids and cracks. If during petrographic examination ettringite is observed in cracks, there is a question as to whether it caused the cracking or merely formed in the available space created through the action of another deterioration mechanism (DePuy 1994). Given that SEF is common in older, hardened concrete, it is at times difficult to attribute it to the observed distress characteristic of paste expansion. In the case where a crack or gap is filled with ettringite, it is often unclear whether the crack or gap came first or did expansion of the ettringite cause the defect. For example, in a recent publication, Erlin (1996b) suggests that the cement paste expansion can result from any number of phenomena including freeze-thaw cycling, hydration of free lime and magnesia, and ASR. Once expansion and cracking occur, secondary ettringite can form and expand into the newly created cracks and voids.
More quantitatively, another researcher has shown using x-ray diffraction, that no ettringite was present in cement mortars immediately after steam curing. However, after the concrete had hardened and a crack structure had formed, primary and secondary ettringite formed to fill the available space (Scrivener 1996). The same researcher also presents reasons why secondary ettringite forms in cracks, rather than creating cracks. The principal reason given is that the level of sulfate saturation in the pore water system of normal concrete does not meet that required to produce a crystallization pressure large enough to cause cracking (Scrivener 1996). The concept of crystallization pressure is also discussed in detail by Johansen et al. (1993) who agree that the necessary conditions for crystallization pressures sufficient to cause cracking in hardened concrete do not exist for the case of SEF.
Ettringite loses a considerable amount of water upon drying (Mindess and Young 1981). Given this fact, swelling of poorly crystalline secondary ettringite due to absorption of water in an alkaline environment is another theory of expansion supported by Mehta (1991) and Wolter (1997). Mehta (1991) cites a study that provided experimental evidence of swelling of microcrystalline ettringite. As previously discussed, Wolter (1997) and Niemann and Lehtonen (1997) suggest that SEF can completely fill the air-void system to the point where it is no longer available to protect the paste from freeze-thaw damage. Thus deterioration occurs due to freeze-thaw action resulting from SEF.
Corrosion of Embedded Steel
Corrosion of embedded steel manifests as scaling, cracking, and associated deterioration of the concrete in areas above or surrounding areas affected by active corrosion. A film of tightly adhering corrosion products normally helps protect reinforcing steel within a concrete structure from continued corrosion (Perenchio 1994). Depassivation of the steel requires that the calcium hydroxide crystals typically present at the steel-cement paste interface are first decomposed and removed. This is followed by the destruction of the passive iron oxide film (Mehta 1991). This film is thermodynamically stable at pH levels greater than 11 (Mehta 1991). Once this film is breached, however, either through a reduction in concrete alkalinity or due to the presence of chloride ions, the deleterious corrosion process can occur. Typically, corrosion due to chloride ingress proceeds at a rate much faster then carbonation due to reduction in alkalinity (e.g. carbonation of the surrounding paste).
The mechanisms involved in the corrosion of steel reinforcement can be identified and explained using basic chemical and physical processes. When any metal corrodes, in essence, it is returning to its natural state as an oxide or hydroxide. Very few metals are found naturally in their metallic state because it is their highest energy state. Since all materials seek to exist at their lowest energy level, the metals will naturally oxidize if possible. This is what drives the corrosion of steel reinforcement (Perenchio 1994). ACI 222R (1989) provides an in-depth description of this corrosion mechanism.
Three components, the anode, the cathode, and the electrolyte, are necessary for a corrosive electrolytic cell to exist. The anode is the site where the metal corrosion occurs through the loss of electrons and oxidization. The anodic process cannot occur until the passive iron oxide (Fe2O3) film is either removed due to lowering of the concrete alkalinity or made permeable in the presence of chloride ions (Mehta and Monteiro 1993). The cathode is the site where the excess electrons from the anode are consumed. It is said that the metal at this site is reduced. The cathodic process requires both oxygen and water to be present at the steel surface (Mehta 1994). Finally, an electrolyte is needed to couple the anode to the cathode. The electrolyte allows the exchange of electrons, and is defined as the medium through which the current that drives the anodic and cathodic reactions flows (Fraczek 1987). The corrosion process is illustrated in figure A-1.
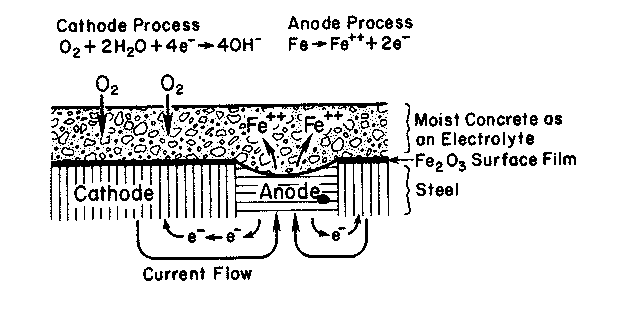
Figure A-1. The electrochemical process of steel corrosion (Mehta and Monteiro
1993).
The following equations describe the electrochemical process involved in steel corrosion (Mehta 1991):
| Anode Reaction: Fe à 2e- + Fe2+ |
| Cathode Reaction: ½O2 + H2O+ 2e- à 2(OH)- |
| Adding Reactions: Fe + ½O2 + H2O à Fe2++ 2(OH)- à iron oxide | (A-13) |
Corrosion of reinforcing steel occurs in concrete as an electrolytic cell is formed between the reinforcing steel and the surrounding concrete. The concrete matrix acts as the electrolyte of the cell by allowing the transfer of electrons between the anode and the cathode. The anode and the cathode of the electrolytic cell may be formed within the structure either on the same dowel, tie, or reinforcing bar or on two separate bars in the concrete. To accomplish this an upper reinforcing bar or dowel will act as the anode, giving up some of its electrons so it can oxidize. A lower bar will act as the cathode having access to oxygen from the bottom of the slab. The reaction product formed from the corrosion of the upper reinforcing steel is iron oxide, commonly known as rust. Rust is responsible for most of the damage caused by corrosion, as its formation is an expansive reaction. Because the reinforcement is tightly embedded within the concrete, the expansion results in cracking and spalling.
Free chloride ions are a major contributing factor in the corrosion of reinforcing steel. Free chloride comes from many sources, including deicing salts, aggregate, cement, mixing water, and chemical admixtures (Perenchio 1994). In the presence of moisture and oxygen, chloride ion concentrations in excess of a threshold level can accelerate corrosion (ACI 1992a). The exact concentration of chloride ions that constitutes this threshold level is debated, but the general consensus is that some chloride ions can be tolerated (ACI 1989). A number of studies cited in ACI 222R (1989) found that corrosion of embedded steel was initiated if the acid-soluble chloride concentration exceeded 0.20 percent. Mehta and Monteiro (1993) suggest the threshold value is in the range of 0.6 to 0.9 kg of chloride ions (Cl–) per cubic meter of concrete. They also note that the hygroscopic nature of chlorides will increase the saturation level of the concrete, further enhancing the corrosion process.
One theory on the effect of chloride ions on reinforcement corrosion states that when the chloride concentration becomes large enough, ferrous chloride, or a ferrous chloride complex, is formed on the steel surface of the reinforcing bars. This ferrous chloride replaces the ferric oxide film that was stabilized by the high pH of the cement paste. Since the chlorides are more soluble than oxides, chloride ions move away from the steel, exposing fresh iron to the now corrosive local environment and instigating the electrochemical cell (Perenchio 1994). Chloride ions alone are insufficient to cause corrosion of reinforcing steel (Mehta 1991); in fact, they act more like a catalyst to the corrosion reaction. Adequate moisture and oxygen must also be available.
Carbonation, a reaction between the calcium hydroxide in the hardened cement paste and the carbon dioxide present in the atmosphere, has both a positive and negative impact on the corrosion reaction. Perenchio (1994) has defined carbonation as atmospheric corrosion of the concrete cover. The reaction produces calcium carbonate and water, reducing the pH of the concrete due to the removal of the hydroxyl ions from the pore water solution (Fraczek 1987). Un-carbonated cement paste has a minimum pH of 12.5, but carbonation can reduce this to levels of 8 to 9 (ACI 1992a). This reduction in the pH of the pore solution results in the removal of the protective passive film coating the reinforcing bars, thus contributing to corrosion. However, carbonation can have a positive effect by decreasing concrete permeability, thereby stemming the flow of moisture, oxygen, and chloride ions, and improving the concrete’s resistance to corrosion of reinforcement (Emmons 1994). Normally, carbonation is not a problem unless the concrete is cracked or the cover is inadequate (ACI 1989).


 ·H 32
·H 32