U.S. Department of Transportation
Federal Highway Administration
1200 New Jersey Avenue, SE
Washington, DC 20590
202-366-4000
Federal Highway Administration Research and Technology
Coordinating, Developing, and Delivering Highway Transportation Innovations
 |
| This report is an archived publication and may contain dated technical, contact, and link information |
|
Publication Number: FHWA-HRT-04-150 Date: July 2006 |
Previous | Table of Contents | Next
The application of the SEM in petrographic analysis of cementitious materials and concrete microstructure (Bentz and Stutzman, 1993; Struble and Stutzman, 1989; Stutzman, 1990, 1991,1993; Stutzman and Bentz, 1993) is becoming increasingly common. SEM imaging provides detailed images of the microstructure that augment those from stereo and optical microscopy. The primary advantages are the high-contrast images of the microstructure, the high spatial resolution of the images, and the ability to perform simultaneous imaging and chemical analysis. This chapter presents an overview of the SEM and includes guidelines for specimen preparation, common imaging methods, and image processing and analysis for quantitative microscopy.
The SEM (figure 173) scans a focused beam of electrons across the specimen and measures any of several signals resulting from the electron beam interaction with the specimen. The most commonly used imaging modes are secondary electron, backscattered electron, and x ray. Images are monochrome since they reflect the electron or x&ray flux resulting from the beam⁄specimen interaction. Computer-based image processing and analysis make routine quantitative imaging possible.
Figure 173. Scanning electron microscope.

Secondary electrons (SE) are low-energy electrons produced as a result of an inelastic collision of a primary beam electron with an electron of an atom within the specimen (Goldstein, et al., 1992). Because of their low energy, they are readily absorbed and only those produced near the surface escape, producing the detailed images of surface topography. The apparent shadowing in the image is a result of the absorption of the SEs by intermediary parts of the specimen. The maximum depth of SE emission is reported to be about 1 nanometer (nm) in metals and 10 nm in insulating materials, depending on the beam accelerating voltage (Goldstein, et al., 1992). Specimens intended for secondary imaging benefit from a conductive coating of a heavy metal such as gold or gold/palladium to increase secondary electron flux, improving the imaging signal. SE images are used to study particle size, shape, surface roughness, and fracture surfaces.
SE images of fracture surfaces of hardened cement paste (figures 174 and 175) show a platy, or foil-like, calcium silicate hydrate (CSH) (figure 174)-slender fibers of calcium silicate hydrate and, along the base of the image, calcium hydroxide (CH) (platy habit). Figure 175 exhibits plates of CH (typical hexagonal habit) and the needle-like habit of ettringite. The existence of the characteristic morphologies depends on the availability of void space for uninhibited growth.
SE imaging is therefore particularly useful in the examination of early-age paste microstructure, high-magnification imaging, examination of secondary crystal growth and fragments of material that may be plucked from a larger specimen, and examination of aggregate texture. Knowledge of the morphological characteristics and bulk chemistries of the hardened cement paste and mineral constituents aids in their identification. Although SE imaging is useful for examining surface texture, the rough surface makes measurements of phase abundance unreliable. As the hardened cement paste matures, filling of the void spaces eliminates the well-formed crystals shown in these figures, with the resulting microstructure appearing to be nondescript. Backscattered electron and x-ray imaging are ultimately more useful in the examination of these microstructures.
Backscattered electrons (BE) are high-energy electrons (typically greater than 50 electronvolts (eV)) that have undergone multiple elastic scattering events within the specimen. The greater energy results in a larger interaction volume and lower spatial resolution compared to the SE image. However, the BE image contrast is generated by the composition of the different phases relative to their average atomic number and is observed by the differential brightness in the image.
Figure 174. SE image showing platy or foil-like CSH, fine bundles of CSH fibers and platy CH (top).
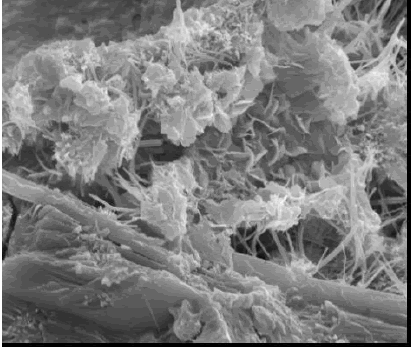
Figure 175. SE image showing platy CH and ettringite needles and also plate-like CH morphology (bottom).

The backscatter coefficient (η) is a measure of the BE fraction for a pure element (Z), which may be estimated using the equation in figure 176 from Goldstein, et al. (1992):
Figure 176. Equation to estimate backscatter coefficient from Goldstein, et al. (1992).
The BE coefficient of a multi-element phase is estimated using the mass fractions (Ci) and ? values for each constituent:
Figure 177. Equation to estimate BE coefficient of a multielement phase.
Contrast between constituents can be calculated as:
Figure 178. Equation to calculate contrast between constituents.
Table 26 lists some phases found in cements and hardened cement pastes, in descending order of their gray-level intensity. Using the contrast equation (figure 178), initial estimates may be made to see if constituents can be distinguished on the basis of their BE intensities. The contrast (6.8 percent) between alite ( ![]() = 15.06) and belite (
= 15.06) and belite ( ![]() = 14.56) is relatively strong, whereas that between belite and cubic aluminate (
= 14.56) is relatively strong, whereas that between belite and cubic aluminate ( ![]() = 14.34) at 1.5 percent is usually too weak to distinguish these constituents. Detector specifications usually quote a 0.5
= 14.34) at 1.5 percent is usually too weak to distinguish these constituents. Detector specifications usually quote a 0.5 ![]() resolution. In difficult situations, a slower scan rate may improve the signal-to-noise ratio to allow distinction. In many of these cases, however, the phases are chemically distinct and the use of x-ray microanalysis will allow their discrimination and identification. Two factors do affect the gray-level intensity of a constituent: (1) porosity within a phase lowers its Z value and will make it appear darker than expected and (2) a sol d solution, if present, will also influence the intensity of a phase proportional to its deviation from these idealized compositions. Examples of these include porosity within CSH for the former and zoning within the ferrite phase for the latter. In general though, anhydrous cement appears brightest, followed by calcium hydroxide, calcium silicate hydrate, and aggregate. Voids appear dark.
resolution. In difficult situations, a slower scan rate may improve the signal-to-noise ratio to allow distinction. In many of these cases, however, the phases are chemically distinct and the use of x-ray microanalysis will allow their discrimination and identification. Two factors do affect the gray-level intensity of a constituent: (1) porosity within a phase lowers its Z value and will make it appear darker than expected and (2) a sol d solution, if present, will also influence the intensity of a phase proportional to its deviation from these idealized compositions. Examples of these include porosity within CSH for the former and zoning within the ferrite phase for the latter. In general though, anhydrous cement appears brightest, followed by calcium hydroxide, calcium silicate hydrate, and aggregate. Voids appear dark.
Figures 179 through 181 provide a comparison of stereomicroscopy and SEM BE and SE imaging. The stereomicroscopic image (figure 179) has a field width of about 26 mm, showing a cross section of pavement concrete exhibiting alkali-aggregate reaction. The SEM image (figure 180) illustrates the same microstructure, but with image contrast based on BE. Figure 181 is another section of the same pavement, but with an SE image of the surface topography. Features such as aggregate, air voids, and fractures are visible in each of the micrographs. The SEM BE and stereomicroscopic images are the most similar as they are both of polished sections.
For both image types, feature identification is made by recognizing a feature as an aggregate, cement paste binder, air void, or fracture. The sand in the SEM image appears as a uniform shade of gray with some brighter grains. The coarse aggregate has a more varied BE signal reflecting the multiphase composition. Identification of the aggregate mineralogy is made in subsequent figures.
| Phase | Composition | η | |
|---|---|---|---|
| Cement Phases | |||
| Ferrite | Ca2(Al,Fe)2O5 | 16.65 | 0.1860 |
| Free lime | CaO | 16.58 | 0.1882 |
| Alite | Ca3SiO5 | 15.06 | 0.1716 |
| Belite | Ca2SiO4 | 14.56 | 0.1662 |
| Arcanite | K2SO4 | 14.41 | 0.1652 |
| Aluminate-cub. | Ca3Al2O6 | 14.34 | 0.1652 |
| Aluminate-ort. | NaCaAl3O9 | 13.87 | 0.1588 |
| Aphthitolite | (Na,K)2SO4 | 13.69 | 0.1577 |
| Syngenite | K2Ca(SO4)2H2O | 13.60 | 0.1556 |
| Anhydrite | CaSO4 | 13.41 | 0.1535 |
| Bassanite | 2CaSO4·H2O | 13.03 | 0.1489 |
| Gypsum | Ca(SO4)·2H2O | 12.12 | 0.1381 |
| Thenardite | Ca(SO4)·2H2O | 10.77 | 0.1249 |
| Periclase | MgO | 10.41 | 0.1213 |
| Hydration and secondary products: | |||
| Portlandite | Ca(OH)2 | 14.30 | 0.1617 |
| Tobermorite | Ca5Si6O18H2 | 13.16 | 0.1539 |
| Hemicarbonate | [Ca2Al(OH)6][0.5 H2O 0.25 CO3 aq] | 12.39 | 0.1406 |
| Hydrogarnet | Ca3Al2(OH)12 | 12.30 | 0.1398 |
| Gypsum | Ca(SO4)·2H2O | 12.12 | 0.1381 |
| Plombierite | Ca5Si6O16(OH)2 8H2O | 12.09 | 0.1380 |
| Monosulfate | [Ca2Al(OH)6][0.5(SO4)3H2O] | 11.66 | 0.1324 |
| Ettringite | {Ca6[Al(OH)6]2·24H2O}[(SO4)3·1.5H2O] | 10.79 | 0.1222 |
| Thaumasite | Ca3Si(OH)6·24H2O (CO3)(SO4) | 9.74 | 0.1097 |
| Brucite | Mg(OH)2 | 9.43 | 0.1085 |
| Hydrotalcite | [Mg0.75Al0.25(OH)2](CO3)0.125(H2O)0.5 | 9.09 | 0.1042 |
| Aggregate phases: | |||
| Calcite | CaCO3 | 12.56 | 0.1422 |
| Dolomite | CaMg(CO3)2 | 10.87 | 0.1237 |
| Quartz | SiO2 | 10.80 | 0.1254 |
The fracture surface (figure 181) at about the same magnification shows the hardened cement paste, aggregates, and some air voids; however, the rough surface makes identification of these constituents more difficult. This mode of SEM imaging is best suited for higher magnification examinations of microstructural features.
The images presented in figures 179 through 184 show that the SEM can work like a stereomicroscope in low-magnification screening of concrete microstructures. It may also be used to examine paste microstructures as seen in figures 185 and 186 with lower and higher magnification images.
Figure 179. Stereomicroscopic image.

Figure 180. Backscattered electron image (same specimen).

Field width is about 15 mm.
Figure 181. Secondary electron image of a fresh-fracture surface from concrete.

Field width is about 15 mm.
Figure 182. Top. Paired image at successively greater magnification of stereomicroscope (white light) images and SEM BE images: Field width is 11 mm.

Figure 183. Middle. Field width is 3 mm.

Figure 184. Bottom. Field width is 1.5 mm.

Figure 185. Backscattered electron image of an epoxy-impregnated, polished cross section of concrete (field width is 2 mm).

a = aggregate, c = residual cement, ch = calcium hydroxide, ip = calcium-silicate-hydrate, inner product, and op = calcium-silicate-hydrate, outer product
Figure 186. Backscattered electron image of an epoxy-impregnated, polished cross section of concrete (field width is 1 mm).

a = aggregate, c = residual cement, ch = calcium hydroxide, ip = calcium-silicate-hydrate, inner product, and op = calcium-silicate-hydrate, outer product
X-radiation is produced when a specimen is bombarded by high-energy electrons. The beam electrons eject inner-shell electrons that are then replaced by outer-shell electrons. This replacement results in emission of an x ray characteristic for that element. The x-ray energy level is displayed as the number of counts at each energy interval and appears as peaks on a continuous background (figure 187), referred to as an energy-dispersive x-ray spectrum (EDX).
Figure 187. EDX spectrum.
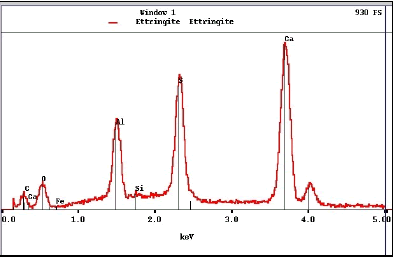
Peak positions are specific to a particular element, intensity (height) is proportional to its relative abundance. This spectrum, from ettringite, exhibits that mineral’s characteristic composition with oxygen, aluminum, sulfur, and calcium. Note that units on the horizontal axis are kiloelectronvolts (keV).
The peaks are designated as k, l, or m. The positions of the peaks are characteristic of a specific element, so phase identifications are made by examination of peak positions and relative intensities (figure 188).
The x-ray signal is used for: (1) spectrum analysis for qualitative and quantitative chemical analyses (figure 188), (2) line scan analysis to plot relative concentration of selected elements along a line (figure 189), (3) x-ray imaging (XR) of element spatial distribution and relative concentrations (figure 190), and (4) an aid in phase identification through its chemical signature. Phase identification is not absolute as in x-ray powder diffraction; however, combining criteria (e.g., BE signal brightness, morphology, associations with other constituents, and chemical composition) provides a rather strong basis for identification. Coupling microscopy with x-ray powder diffraction data is also useful as it can be used to limit the elements in the potential compounds to those present in the specimen. Mass concentration to a few tenths of a percent can be detected using an energy dispersive x-ray detector. The relative accuracy of quantitative analysis (using certified standards) is about ±20 perce t for concentrations of about 1 percent, and ±2 percent for concentrations greater than 50 percent. More details on x-ray microanalysis may be found in Goldstein, et al. (1992).
Figure 188. EDX spectra for ettringite (top left), monosulfate (top right), calcium hydroxide (bottom left), and calcium-silicate-hydrate (bottom right) aid in phase identification characteristics.
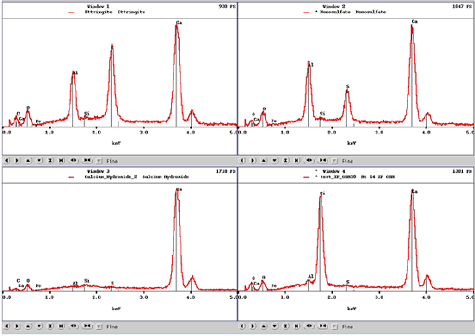
The peaks and their relative intensities are affected by the accelerating voltage of the microscope beam (figure 191). If the voltage is too low, then the beam energy may not be capable of generating the characteristic radiation. For example, the iron K line occurs at 6.4 keV and requires about 11 keV for adequate excitation. Accelerating voltages between 12 and 15 keV provide for reasonable BE and x-ray imaging. Lower accelerating voltages decrease the interaction volume, increasing resolution, but with a loss in efficiency in generating higher energy x rays.
Lower beam energies are not as efficient in exciting high-energy x rays, and the lower x-ray absorption accentuates the low-energy x rays. High beam energies result in greater absorption in the lower energy range. For cements, an accelerating voltage of 12 to 15 keV balances the need for adequate beam energy without excessive absorption of lower energy x rays. Higher beam energies are favored for SE imaging as they result in increased depth of field and smaller beam diameters and, thus, greater resolution.
Figure 189. Fence diagram created by collecting a series of EDX spectra along an emplacement of ASR gel (the gel composition rapidly becomes much like the bulk CSH once outside the aggregate and the source of reaction).
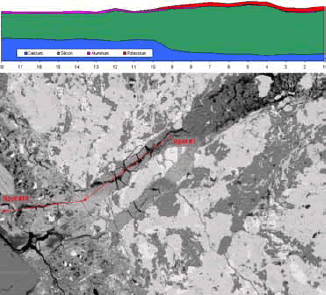
Figure 190. Plots of element spatial distribution are generated through x-ray imaging.
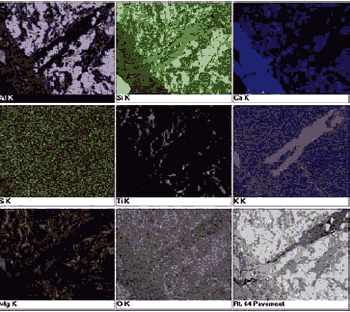
Figure 191. Effects of different accelerating voltage on x-ray spectra of ettringite show the loss of low-energy peaks at high accelerating voltages and the opposite effect at low voltages.
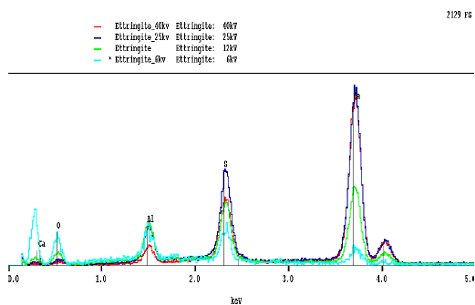
X-ray spectra (low to high energy, keV, left to right) for different accelerating voltages. Note that, in this case, accelerating voltages are given as "kv," which are the same as keV, as discussed earlier.
The SEM may be used to examine portland cement clinker, cement powder, cement pastes, mortars, and hardened portland cement concrete. Specimen preparation uses an epoxyimpregnated, polished section where the epoxy resin permeates the material’s pore system or encases powder particles. The specimens are then cut or ground to expose a fresh surface, and that surface is then polished using a series of successively finer grades of diamond paste. This polishing stage is necessary to remove cutting and grinding damage, exposing an unaltered cross section of the material’s microstructure. Polished thin sections prepared as described in section 5.3.4 can be examined both with transmitted light and with the SEM (see figures 121 through 123). Sahu, et al. (2001) described coupled transmitted-light and SEM examinations of thin sections.
Epoxy impregnation of the pore system serves two purposes: (1) it fills the voids and, upon curing, supports the microstructure, serving to restrain it against shrinkage cracking, and (2) it enhances contrast among the pores, hydration products, and cementitious material. With relatively high-permeability materials or powders such as clinker or portland cement, an epoxy of low viscosity is necessary, whereas for the less-permeable cement pastes and concretes, an ultra-low viscosity epoxy aids in rapid infiltration of the pore structure. Examples of epoxies that are used for impregnation/embedment are LR White (hard grade) (ultra-low viscosity) and EPOTEK 301 (medium low viscosity) (chapter 2); other suitable materials may be used.
BE and x-ray imaging requires a highly polished surface for optimum imaging and x-ray microanalysis (Stutzman and Clifton, 1999). Rough-textured surfaces diminish the image quality by reducing contrast and loss of feature definition. In addition, the lack of a polished specimen makes quantitative estimates arduous, as the surface is no longer planar. Sawcut surfaces that have not been epoxy-impregnated, or polished, do not present the true microstructure and are difficult to examine and interpret without bias. The sawing operation damages the microstructure through the creation of a series of fractures, which are then enhanced with subsequent drying shrinkage. For SE imaging, the saw damage dominates the topographic features, and particulate matter deposited on the surface may be mistaken for undisturbed material. Polished, epoxyimpregnated specimens are relatively simple to prepare and the above-mentioned difficulties can be avoided.
A comparison of BE images of a sawcut surface (figure 192) with an epoxy-impregnated, polished surface (figure 193) of the same concrete core illustrates the marked differences in feature clarity and artifacts. In figure 192, the sawcut surface exhibits cracking that resulted from drying shrinkage of a surface-damaged specimen. The surface, being rough and partly covered with particulate matter, exhibits little phase-related contrast. The image shows an aggregate at the base; however, aside from the uniform hardened paste, no distinct hydration products can be discerned. The epoxy-impregnated, polished specimen (figure 193) shows microstructural feature clarity not seen in the images in figure 192. Here, the aggregate is not only clearly seen, but one can also distinguish between siliceous and carbonate aggregate by gray level. The residual cement grains appear bright, and large voids within both the paste and the aggregate are dark. In the higher magnification image (bottom), the hardened cement paste/ aggregate interfacial zone is shown. The residual cement grains are the brightest feature, followed by CH, carbonate aggregate, CSH, and finally the black pores. A highly polished surface also aids in x-ray imaging. Figure 194 illustrates the use of x-ray imaging of a region shown in the BE image (top) to examine element spatial distribution (bottom). In this example, visual assessment of calcium, aluminum, and sulfur images allow one to locate monosulfate within the hardened cement paste. Referring back to the BE image shows the monosulfate as a uniform gray that is slightly darker than CSH, with a platy parting.
Figure 192. BE images of unimpregnated, sawed surface (the sawed surface is rough with residual particulate matter, yielding poor contrast and shadowing that make interpretation of BE and EDX images difficult).

Figure 193. BE images of the impregnated, polished surface from the same concrete (polished surface presents optimum BE and EDX imaging characteristics).

Figure 194. Polished section of hardened portland cement paste imaged using the BE signal clearly shows the constituent phases (residual cement (RC) appears brightest, followed by CH, CSH, and other hydration products) (top); EDX image (bottom) shows regions of intermediate-intensity calcium (blue), intermediate-intensity aluminum (purple), and highintensity sulfur (yellow), defining locations of monosulfate that could not be distinguished on a rough surface (field width is 73 µm).

A list of equipment and materials necessary for the preparation of polished specimens is given in table 27. For some items, substitution may be possible if comparable supplies are available in the laboratory. The list is presented in order of use of the equipment or supplies.
| Item | Purpose |
|---|---|
| Diamond blade slab saw | Large-sized sample slabbing |
| Diamond blade wafering saw | Cutting of thin (millimeter-sized) sections |
| Propylene glycol | Diamond-saw cutting lubricant |
| Alcohol: 200-proof ethanol | Cutting lubricant, cleaning aid |
| Ultrasonic bath | Specimen cleaning |
| Specimen jars and lids | For replacement steps |
| Potting epoxies (medium and low viscosity) | For powders and hardened pastes |
| Dye, blue or red, alcohol-miscible | LTo estimate alcohol replacement depth |
| Refrigerator | Epoxy storage |
| Vacuum chamber and pump | Vacuum impregnation |
| Drying/curing oven | Capable of at least 65 °C |
| Glass plate (400 by 400 mm) | Smooth surface for grinding |
| Lapidary wheel (minimum 200 mm) | Grinding and polishing |
| Mold cups | Potting specimens |
| Aluminum foil (extra heavy duty) | For forming odd-sized specimen molds |
| Mold release | Facilitates removal of specimen/epoxy |
| Metal trays to hold specimens | Contains any leaking epoxy |
| Diamond pen | Label engraving |
| Abrasive papers (silicon carbide) | Coarse to fine grinding, 100 to 600 grit |
| Polishing cloths (low-relief) | 6µm and finer polishing |
| Diamond paste for polishing | 6, 3, 1, 0.25 µm in non-aqueous suspension |
| Lint-free cloths | Specimen handling and cleaning |
| Compressed air | Specimen cleaning and drying |
| Vacuum desiccators | Specimen storage |
Powder mounts are prepared by suspending cement powder in epoxy, curing the epoxy, and cutting and polishing a surface of the powder/epoxy composite. The cement powder is mixed in about 5 grams (g) of epoxy, using enough cement to form a stiff ball. The cement/epoxy mixture is placed in a mold container and pressed to fill the base of the mold. The mixture is then consolidated in the sample mold by sharply tapping it on the laboratory bench top, and the epoxy is cured in accordance with the manufacturer&s guidelines. After curing, the specimen is removed from the mold and labeled and a fresh surface is exposed using a wafering saw or by grinding. Examples of this application may be found in Struble and Stutzman (1989) and Stutzman (1990).
Cement pastes, mortars, and concretes may be prepared in two ways: (1) dry potting and (2) wet potting. Dry potting is used when the specimen has been dried before, when drying shrinkagerelated cracking is not of concern, or when rapid preparation is needed. Wet potting is used to prepare a polished section where the material has not been dried and therefore has not undergone any drying shrinkage. Cracks observed in wet-potted specimens may then be ascribed to physical or chemical processes acting upon the concrete prior to specimen preparation, and not to dryingrelated shrinkage resulting from preparation techniques.
Dry-specimen potting involves taking a sawed section or block of material and drying the specimen at a low temperature (less than 65 °C). Removal of water is necessary as it can interfere with polymerization of the epoxy. The specimen is then placed in a container and surrounded by epoxy, leaving the top surface exposed to laboratory air, allowing the lowviscosity epoxy to be drawn into the microstructure by capillary suction. To speed infiltration, or when using a medium-viscosity epoxy, the specimen may be immersed in epoxy and a vacuum drawn to remove the remaining air. Vacuum-impregnation procedures are described in chapter 5 (section 5.3.4.2). Upon release of the vacuum, the epoxy is forced into the pore system. The epoxy is cured at a low temperature (65 °C) and is then ready for cutting and polishing.
Wet-specimen potting is a three-step process: (1) the pore solution is replaced with alcohol (200-proof ethanol), (2) the ethanol is replaced with a low-viscosity epoxy, and (3) the epoxy is then cured. The slab and wafering saws are lubricated with propylene glycol or isopropyl alcohol to keep the specimen from drying when cutting. The cut section is then placed in a lidded jar filled with 200-proof ethanol for the alcohol-pore solution replacement stage. The use of a companion specimen allows one to gauge the time necessary for the alcohol-pore solution replacement. This companion specimen is usually a remnant from the specimen after trimming. This specimen is placed in a jar filled with ethanol dyed a deep red or blue using any alcoholmiscible dye. By splitting or sawing the companion specimen after a period of time, the depth of replacement can be observed by the depth of dye coloration. When this front is equal to half the section thickness, the pore solution in the section has been replaced by alchol. The section is then placed in a container with the low-viscosity epoxy. The time necessary for epoxy replacement of the alcohol is at least equal to that required for the first replacement stage. In the NIST laboratory, we allow about 1.5 times the pore solution-alcohol replacement time for that of the epoxy-alcohol replacement. Implicit in this method is that the thinner the section, the shorter the time that is required for each stage. The specimen is placed in a mold with fresh epoxy, which is then cured at low temperatures according to the manufacturer’s directions. The specimen is then ready for the cutting and polishing stages.
The cutting, grinding, and polishing steps are common to all preparations in order to expose a fresh surface. Diamond-blade slab or wafering saws, lubricated using propylene glycol, are suitable for exposing a fresh surface. This surface needs to be smoothed by grinding. Abrasive papers of 220, 320, 400, and 600 grit (silicon carbide paper) used dry are also suitable for rapid removal of material by grinding. VTRC uses diamond-impregnated lapping films (chapter 2) (available in 125, 75, 45, 30, 15, 9, and 6 µm) at this stage. Using successively finer grades of abrasive paper removes damage produced by the earlier step. After grinding with the 600 grit paper, the surface is smooth enough for polishing with the diamond pastes. Visual examination of the specimen allows one to identify when the abrasive has cut the entire surface. Grinding striations on the specimen surface indicate that the grit has removed a layer of material. By alternating grinding directions by 90 degrees, one can ensure that the entire surface has been ground. These operations damage the specimen surface, necessitating a polishing step that is described next.
Polishing removes the damage imparted by the sawing and grinding operations (figures 192 and 193). This stage involves use of a sequence of successively finer particle-size diamond polishing pastes ranging from 6 to 0.25 µm, and a lap wheel covered with a low-relief polishing cloth. This may be performed manually or, for greater sample throughput, a semi-automated polisher may be used.
Variable-pressure electron microscopes are operable with excellent results when using uncoated specimens. For those systems that require a high vacuum, a thin coating of carbon serves to dissipate excess charge from the specimen while exhibiting little effect on image contrast and little interference with elements of interest. Metal coatings such as gold or gold palladium are suitable for secondary imaging of topographic features. However, their x-ray lines interfere with elements of interest in cementitious materials (sulfur) and the heavy metal decreases BE contrast.
The various SEM examination modes are discussed in section 14.2. That information, together with the specific issues regarding the study at hand, guided mode selection. For general concrete characterization and investigation of materials-related distress, the coupling of information gained through BE and EDX examination modes has been found most useful. Review information in chapters 3, 8, and 10 through 13 and Van Dam (2002b) regarding the planning of examinations. Typical settings for routine examinations in the three basic modes are outlined in table 28.
Examples of the use of BE and EDX techniques are given in sections 14.2.2 and 14.2.3. Examples of their use in investigating AAR are shown in chapter 10. The high resolution and the ability to perform chemical analyses make these modes ideal for examining HCC paste. The hydration products appear as an intermediate gray, and the residual cement appears to be bright. The aggregate may be recognized using both its gray level and uniformity and its shapes. Silicates such as quartz appear to be slightly darker than the cement paste (figures 185 and 186), whereas feldspars can be distinguished by the presence of Ca, or Na and K. Calcium carbonate aggregate appears similar in gray level to that of the paste; dolomite can be distinguished from calcite by its slightly darker gray level and the presence of Mg (figure 196). In differentiating carbonate aggregates from hydration products, the feature&s uniformity and shape are characteristics that are useful in distinguishing aggregates from the hardened paste atrix (figures 197 and 198).
Close examination of the microstructure of a 28-day hardened portland cement paste is demonstrated in figure 194 in which BE and EDX images display constituents of the paste.
| Secondary electron imaging (SEI) |
|
| Backscattered electron imaging (BEI) |
|
| X-ray imaging/x-ray microanalysis(EDX) |
|
For cement pastes, residual cement grains appear brightest, followed by the hydration products CH, CSH, and the darkest, being the epoxy-filled pore spaces (review table 26). Other constituents such as ettringite and monosulfate appear similar in gray level to that of CSH and require x-ray analysis for location and identification. Both of these phases do show greater uniformity and perhaps a slightly darker gray than that of CSH, and they also exhibit a platy parting that is, in part, a result of desiccation. X-ray imaging (figure 194, lower images) demonstrates how examination for regions of intermediate-intensity calcium (blue), intermediate-intensity aluminum (purple), and high-intensity sulfur (yellow) define locations of monosulfoaluminate.
For the cement grain (figure 194), the ferrite phase appears brightest in the BE (upper) image, followed by alite, belite, aluminate, and periclase. X-ray imaging facilitates identification of periclase, alkali sulfates, and calcium sulfates. Figures 198 and 199 show that for a 28-day hardened cement paste microstructure, polishing yields clear definition of the constituents?the black pore space filled with the cured epoxy, the bright grains of residual cement (C), the intermediate-gray CSH, and the somewhat brighter gray CH.
The relationship between area fraction and volume fraction has been recognized for a long time and was mathematically derived by Chayes (1956).
There are three established methods for determining the area percentage of a phase in a planar section (also review section 6.3.2): (1) the Delesse method of tracing and weighing each phase group, (2) the Rosiwal-Shand linear traverse method, and (3) the Glagolev-Chayes point-count method.
The Glagolev-Chayes method, or point-count method, is perhaps the most widely used technique in quantitative mineralogical analysis using a microscope. As the name implies, this technique involves sampling the microstructure using a grid of points and identifying the phase falling underneath each point. The rule for selecting a grid, magnification, and crystal size is to select a point spacing slightly greater than the crystal size (i.e., adjacent points should not fall on the same crystal). Additional points are measured by moving the specimen to a new field of view and again counting the phases under the points. The point-count method is relatively simple, yet provides a reliable means to measure phase abundance physically, using a direct measure of the microstructure.
Three principal sources of error were identified by Hofmänner (1975) for consideration when point counting:
For these exercises, it is assumed that a representative sample was collected and that the sample size was reduced using proper practices. The measurement error of the analysis is related to the total number of points counted, with the absolute error at the 96-percent confidence interval given using the equation in figure 195:
Figure 195. Equation for percent error in the point-count method (at the 96-percent confidence level) based on the the proportion of points on the phase of interest and the total number of all points counted.
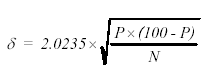
where:
η = absolute error in percent
P = percentage of points of a phase
N = total of all points
The total error becomes smaller as the number of points counted increases; therefore, the greater the number of points counted, the more reliable the measurement. An analysis of 4000 points should provide acceptable precision for phase abundance analysis.
Mass percentages may be calculated by multiplying the volume fractions by the specific gravity of the corresponding clinker phase and normalizing the totals to 100 percent.
In addition to phase abundance, measurement of crystal size, shape, and other parameters may be recorded.
Figure 196. BE image (right) of carbonate aggregate composed of darker gray dolomite (1) rhombs in fine-grained matrix containing lighter gray calcite (6) (the gray-level intensity of calcite and dolomite is distinct, and they can also be distinguished on the basis of the EDX spectra (left); field width is approximately 100 µm).
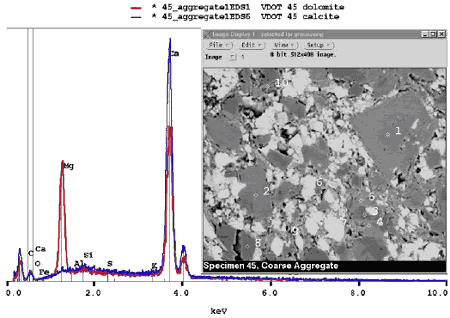
Figure 197. BE images of carbonate aggregate in concrete at 15X (field width of 8 mm).

Aggregate particle is to upper left in left image and lower right in right image. Uniformity and shape are useful in distinguishing carbonate aggregate from a mortar matrix that contains phases of similar gray-level intensity.
Figure 198. BE images of carbonate aggregate in concrete at 400X (field width of 300 µm).
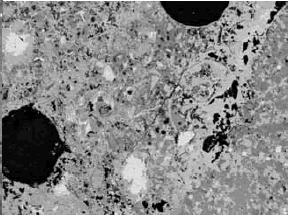
Aggregate particle is to upper left in left image and lower right in right image. Uniformity and shape are useful in distinguishing carbonate aggregate from a mortar matrix that contains phases of similar gray- level intensity.
Computer-based image processing and analysis may also be used to extract information from images, or combinations of images. The BE and x-ray images of polished sections are best suited for this approach as they use the planar section necessary for measurements. Features may be identified on the basis of their gray level (figures 199 and 200) or through combinations of features from images, making use of both x-ray and BE information (figure 201). Identification and measurement of residual cement particles in hydrated cement can be accomplished by taking advantage of the cement particles& bright appearance in the BE image. They may be isolated on the BE brightness alone. Similarly, coarse porosity appears black in the BE image and may also be isolated and measured on this basis.
Figure 202 illustrates the use of BE and EDX images to calculate the percentage of CH in the paste. Information from the analyses similar to those shown in figures 199 through 202 provide data on unhydrated cement, CH, and paste porosity. As noted in chapter 9, these parameters can be used to form a comparative estimate of w/cm. Badger, et al. (2001) reports on quantifying porosity by means of contrast in BE images to estimate w/cm.
A more difficult feature would be mapping the distribution of monosulfate in a hydrated cement paste. The BE coefficient is not such that it is easily distinguishable from CSH. It does exhibit a greater uniformity and, occasionally, platy parting. However, it is compositionally distinct, composed of calcium, aluminum, and sulfur, and may be both identified and mapped using both the BE and x-ray images (figure 194) and quantified as described here.
Figure 199. Gray-level histogram of hardened cement paste.
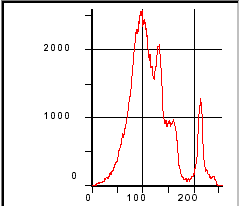
Figure 200. BE image of hardened cement paste (field width is 73 µm).
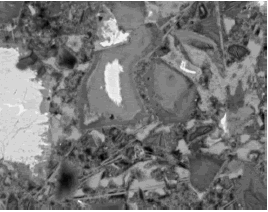
Figure 201. Pseudocolored image based on BE gray scale of figures 199 and 200 (red = residual cement, blue = CH, black = CSH, and green = coarse porosity).Counting the number of pixels for each color allows an estimate of the phase area fractions for this field of view.
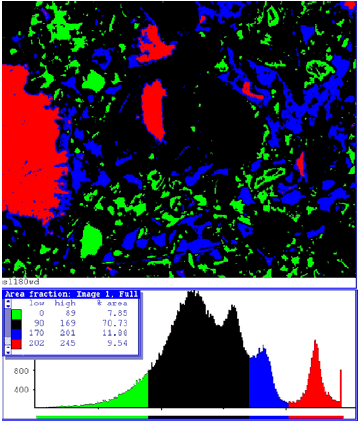
Figure 202. Processing of BE and calcium x-ray images here highlights the CH location (analysis of the binary image of CH distribution calculated 12 percent of the image field to be occupied by CH).

Proper specimen preparation is necessary when using the SEM for the study of cementitious materials and hardened HCC. Rough surfaces, such as those prepared using only fracture, sawcut, or rough-lapped preparations, are not suitable. These preparations may actually create preparation-induced artifacts that are not representative of the microstructure; create a surface that is difficult to describe; and not being planar, are unsuitable for estimates of phase abundance. The BE and x-ray imaging modes are particularly sensitive to rough surfaces, which affect the definition of the constituents through loss of contrast and signal shadowing.
Comparison of SEM and optical microscopic examinations of HCC show that the important features are recognizable in both technologies. The much higher resolving power of the SEM, together with the elemental analysis capabilities of the EDX microprobe, creates a tool that greatly expands our capabilities to study the microstructure of HCC, including the interaction between the various cementitious materials being used today and the microstructural effects of deterioration mechanisms.

